IJCRR - 5(4), February, 2013
Pages: 97-102
Date of Publication: 28-Feb-2013
Print Article
Download XML Download PDF
SINONASAL TERATOCARCINOSARCOMA : A RARE NEOPLASM
Author: Nikunj V. Mehta, Sanjay V. Dhotre, R.N.Gonsai, Tarang B. Kadam, Kalpana K. Dave, Seema K. Modh
Category: Healthcare
Abstract:Background: Sino-nasal teratocarcinosarcoma (SNTCS) is rare and unfamiliar entity with grave prognosis. It was first described by Shanmugaratnam et al. in 1983 and was aptly termed as \"Teratocarcinosarcoma\" by Hefner and Hyams in 1984. As the name suggests, it is composed of benign and malignant epithelial components as well as mesenchymal and neural components Because of its rarity, these lesions are often misdiagnosed as immature teratoma or carcinosarcoma may lead to management difficulties. Objective: The objective is to make pathologists aware with this rare entity and always consider it as differential diagnosis of teratoma and carcinosarcoma which are more common lesions in this location / region. Research Methodology: We report a 30 years old female with SNTCS involving right nasal cavity extending into nasopharynx, maxillary, ethmoid, sphenoid and frontal sinuses. She presented with complains of the nasal blockage, difficulty in breathing and bleeding from right nose since 1 month. Excision biopsy was done. Result: Multiple sections have been taken and final diagnosis of Sino-nasal teratocarcinosarcoma was made.
Keywords: Sino-nasal teratocarcinosarcoma, Malignant.
Full Text:
INTRODUCTION
Sino-nasal teratocarcinosarcoma (SNTCS) is a very rare malignant neoplasm characterized by combined features of immature or malignant teratoma and carcinosarcoma [1,2] . They were previously reported as teratoid carcinoma, malignant teratoma, blastomatous tumours, and blastoma. SNTCS is highly malignant tumour with rapid aggressive growth. Prognosis is poor [1,2]. It was first described by Shanmugaratnam et al. in 1983 and was aptly termed as “Teratocarcinosarcoma” by Hefner and Hyams in 1984. [3,4] . The most common clinical presentation is nasal obstruction and epistaxis.[5,6] It is more common in male with male to female ratio of 7:1[7] . Mean survival time was about 1.7 years after diagnosis with 60% mortality within 3 years[8] . Metastasis is rare and may involve spinal axis, cervical lymph nodes, and respiratory tract [6] .
CASE REPORT
A 30-year-old female presented with nasal obstruction, difficulty in breathing and bleeding from nose since one month. Her hemoglobin level was 13.0 gm/dl at the time of presentation. Other routine hematological and biochemical investigations were normal.
A computed tomography (CT) PNS scan
revealed polypoidal soft tissue mass involving right nasal cavity extending into posterior part of nasal cavity and nasopharynx and into right maxillary sinus with erosion of medial wall of right maxillary sinus ( Fig. I ). A polypoidal mucosal thickening is seen in right maxillary, ethmoid, sphenoid, frontal sinus. Right spheno-ethmoidal recess and fronto-ethmoidal recess appears blocked. Left spheno-ethmoidal recess and fronto-ethmoidal recess appears normal. The mass was in the posterior nasal cavity and nasopharyngeal wall extending into right ethmoidal sinus. The patient was operated with laryngoscopic excision procedure.
HISTOPATHOLOGICAL EXAMINATION
Grossly Single gray–white irregular nodular soft tissue structure measuring 3.5×3×1 cm3 . Cut surface is White with heterogenous consistency
Microscopically:- Sections show tumour tissue consist of cellular, heterogenic tissue elements embedded in proliferative fibrocollagenous stroma. There is presence of primitive looking blastema like hyperchromatic mitotically active densely cellular areas (Fig. IV). There are also definite glandular differentiations with presence of primitive looking glands (Fig. III). Chondromyxoid nodular areas, areas of osseous metaplasia, primitive osteoid tissue lined by pleomorphic tumour cells-tumour osteoid tissue and neuroglial tissue are also seen (Fig. II, V). The stroma between heterologous elements is highly cellular, running in fascicles and mitotically active.
DISCUSSION
Malignant tumours having teratoma and carcinoma were reported previously as teratoid carcinoma, malignant teratoma, or blastematous tumours [9] Heffner and Hyams in 1984 first suggested the tumour to be called as teratocarcinosarcoma in order to describe the complex histological pattern of these neoplasms [4] . The most common clinical presentation for sinonasal teratocarcinoma is nasal obstruction and epistaxis with the average duration of symptoms being reported at 3.5 months[5,6] .Overall, there is a strong male predominance with male to female ratio is 7:1 [7] . Mean survival for this neoplasm has been reported at 1.7 years with a 60% mortality rate within 3 years[8] . Diagnosis of SNTCS can prove to be difficult if only a small incisional biopsy is taken because of the heterogeneity and variegated histological architecture[6] Small sample sizes can underestimate the true histology of this neoplasm and lead to a misdiagnosis such as malignant craniopharyngioma, adenocarcinoma, synovial sarcoma[10] . Histologically, it is different from true carcinosarcoma which consists of a single malignant epithelial and a single malignant mesenchymal component, whereas SNTCS has one or many epithelial and mesenchymal components (both benign and malignant)[11] Variegated architecture and tissue heterogeneity are characteristics of this malignant neoplasm. The malignant epithelial component includes squamous cell carcinoma and adenocarcinoma. “Fetal-type” clear cells, squamous epithelium, and immature neuroepithelium represent important histologic characteristics useful in diagnosis[3,17] . These highly malignant tumours initially present with relatively benign complaints of recurrent epistaxis (53.52% of cases) and nasal obstruction (61.97% of cases). Other manifestations raising suspicions of malignancy, such as odynophagia, dysphagia, expectoration of tissue, epiphora, headache,vision loss, exophthalmos, anosmia and altered mental status arise when the tumour invades surrounding tissues and the severity if related to the degree of tumour extension[12] . The present case had characteristic features of SNTCS including epithelial and mesenchymal elements. Despite several studies, the histogenesis of this tumour remains controversial. Hefner and Hyams postulated that the tumour originates from olfactory membrane due to presence of neural tissue. Some authors believe that SNTCS probably originates from primitive embryonic tissue or immature pleuripotential cells[13] A histogenetic origin from a multipotential adult somatic stem cell with divergent differentiation has been favored over a germ cell origin. This assumption has been based on the lack of germ cell elements and, until recently, the absence of demonstrable amplification of 12p in tumour cells[14] Ultrastructurally, the primitive cells had many neural processes with parallel microtubules. Tumour cells showing squamous cell differentiation were characterized by desmosome-like junction and intracellular tonoflaments. Some of the stromal spindle cells had actin filaments with dense patches and dense core granules[15] In a study by Budrukkar et al., disease recurred in 11 out of 14 patients, with a median time to recurrence of seven months. Multimodality treatment, in the form of a combination of surgery, radiation therapy, and chemotherapy, appears to be the optimal approach [16] A combination of radiotherapy and surgical treatment offers the best five-year survival rate (50%); followed by only surgical treatment (47%). In the recurrent or metastasis lesion, adjuvant chemotherapy may improve the survival rate since the metastatic tissue often contains sarcomata components. Currently no management guidelines are available regarding the disease and most of the literature focus on the histopathological findings. The high rate of recurrence is suggestive of the aggressive biological nature of the disease and although the prognosis does not appear encouraging aggressive surgery followed by chemo-radiation appears to be the mainstay of management for this condition.
Conflict of interest: Authors have declared no conflict of interest.
References:
1. Chao KK, Eng TY, Barnes J, Dahlia R. Sino nasal teratocarcinosarcoma. Am J Oncol 2004;27:29-32.
2. Ogawa T, Ikeda K, Watanabe M, Satake M, Oshima T, Suzuki N, et al. A case report of sinonasal teratocarcinosarcoma. Tohoku J Exp Med 2000;190:51-9.
3. Shanmugaratnam K, Kunaratnam N, Chia KB, Chiang GS, Sinniah R. Teratoid carcinosarcoma of the paranasal sinuses . Pathology 1983;15:413-9.
4. Heffner DK, Hyams VJ. Teratocarcinosarcoma (malignant teratoma?) of the nasal cavity and paranasal sinuses. A clinicipathological study of 20 cases. Cancer 1984;53:2140-54.
5. Salem F, Rosenblum M K, Jhanwar S C, Kancherla P, Ghossein R A, Carlson D L. Teratocarcinosarcoma of the nasal cavity and paranasal sinuses: report of 3 cases with assessment for chromosome 12p status. Hum Pathol. 2008;39(4):605–609.
6. Wellman M, Kerr P D, Battistuzzi S, Cristante L. Paranasal sinus teratocarcinosarcoma with intradural extension. J Otolaryngol/ 2002, 31(3):173- 176.
7. Wei S, Carroll W, Lazenby A, Bell W, Lopez R, Said-Al-Naief N. Sinonasal teratocarcinosarcoma: report of a case with review of literature and treatment outcome. Ann Diagn Pathol.2008;12(6):415–425.
8. Carrizo F, Pineda-Daboin K, Neto A G, Luna M A. Pharyngeal teratocarcinosarcoma: review of the literature and report of two cases. Ann Diagn Pathol. 2006;10(6):339–342.
9. Patchefsky A, Sundmaker W, Marden PA. Malignant teratoma of the ethmoidal sinus. Cancer 1968;21:714-21.
10. Szudek J, Bullock M, Taylor S M. Sinonasal teratocarcinosarcoma involving the cavernous sinus. J Otolaryngol. 2005; 34(4):286–288.
11. Shindo ML, Stanley RB, Kiyabu MT. Carcinosarcoma of the nasal cavity andparanasal sinuses. Head Neck 1990; 12:516- 9.
12. Smith SL, Hessel AC, Luna MA, Malpica A, Rosenthal DI, El-Naggar AK.Sinonasal teratocarcinosarcoma of the head and neck: a report of 10 patients treated at a single institution and comparison with reported series. Arch Otolaryngol Head Neck Surg 2008; 134: 592-5.
13. Su YY, Friedman M, Huang CC, Wilson M, Lin HC. Sinonasal teratocarconosarcoma. Am J Otolaryngol 2010;31:300-3.
14. Thomas J, Adegboyega P, Iloabachie K, Mooring JW, Lian T. Sinonasal teratocarcinosarcoma with yolk sac elements: a neoplasm of somatic or germ cell origin? Ann Diagn Pathol 2011;15:135- 9
15. Shimazaki H, Aida S, Tamai S, Miyazawa T, Nakanobou M. Sinonasal teratocarcinosarcoma: ultrastructural and immunohistochemical evidence of neuroectodermal origin. Ultrastruct Pathol 2000;24:115-22.
16. Budrukkar A, Agarwal JP, Kane S, Siddha M, Laskar SG, Pai P, et al. Management and clinical outcome of sinonasal teratocarcinosarcoma: single institution experience. J Laryngol Otol 2010;124:739- 43.
17. Wang SY, Zhu L, Li SM, Lin L, Zheng SX, Wu YF, et al. Sinonasal teratocarcinosarcoma : a clinical, radiologic and pathologiac study of 5 cases. Zhonghua Bing Li Xue Za Zhi 2007;36:534-8.
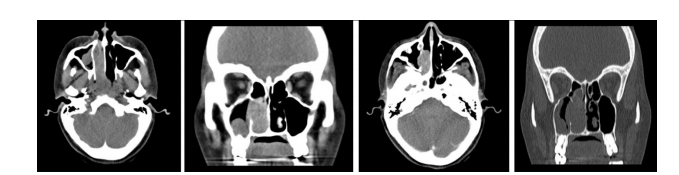
Figure I :- CT Scan of paranasal sinus axial, coronal, bony window and sagittal views showing soft tissue density mass lesion involving right ethmoid sinus extending up to posterior part of nasal cavity and causes erosion of medial wall of right maxillary sinus.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





