IJCRR - 5(19), October, 2013
Pages: 69-80
Date of Publication: 19-Oct-2013
Print Article
Download XML Download PDF
PHYSIOLOGICAL JAUNDICE: ROLE IN OXIDATIVE STRESS
Author: Nitin Pandey, Sushma Gupta, Raj Kumar Yadav, Kumar Sarvottam
Category: Healthcare
Abstract:Physiological jaundice is a common condition encountered in almost two third of neonates. It occurs due to complex interaction of many factors. In this review we have discussed mainly the physiological basis for its development. In newborns, if bilirubin level is more than physiological level, it may cause bilirubin encephalopathy (kernicterus), a deleterious neurological outcome. Then why nature has selected jaundice, a common condition in newborns. Perhaps nature has tried to use the antioxidant property of bilirubin and biliverdin to protect newborns that face storm of oxidative stress after birth.
Keywords: Physiological jaundice, neonates
Full Text:
INTRODUCTION
Jaundice is the visible manifestation in skin and sclera of elevated serum concentrations of bilirubin. Adult jaundice is generally a pathological consequence. Most adults are jaundiced when serum total bilirubin levels exceed 2.0 mg/dL. In neonates, the common causes of jaundice include hepatic immaturity, red cell incompatibility, infection, and breast feeding while some not so common causes are hypothyroidism, galactosaemia, viral hepatitis, and atresia of the bile ducts. The jaundice due to red cell incompatibility appears within 24 hours of birth, and is attributed to incompatible rhesus grouping and incompatible ABO grouping. The infective jaundice is a result of septicemia and urinary tract infection, and is suspected if the jaundice appears after the fourth day of life, though it may have other presentation as well. The jaundice due to hepatic immaturity is termed as physiological jaundice, and is reported in approximately 60% of normal full term infants and in 80% of the preterm infants. Infants, however, may not appear jaundiced until the serum total bilirubin concentration exceeds 5.0 to 7.0 mg/dL. The bilirubin levels gradually increase and reach peak by day 5-7, which can be as high as 12 mg/dL (moderate jaundice) in normal full term infants and up to 14 mg/dL (severe jaundice) in normal premature infants (1). Although neonatal jaundice is harmless, newborns are generally monitored for hyperbilirubinemia, acute bilirubin encephalopathy or kernicterus. However, onset of jaundice within first 24 hours of life, an increase of >5 mg/dL per day, direct bilirubin level > 1 mg/dL at any given time, or the persistence or new onset of jaundice in infants 2 weeks of older is warranted, and should be clinically investigated, especially in breast fed infants (2).
Causes and Presentation
As early as 1925, the animal studies at Rockfeller Institute indicated that the jaundice that develops after obstruction of common duct in the absence of complications, expresses the physiological wastage of corpuscles occurring from day to day (3). Later studies also indicated that physiologic hyperbilirubinemia in the neonate includes an increased bilirubin load because of relative polycythemia, wherein erythrocyte life span have
a shorter lifespan of 80 days compared with the adult erythrocyte life span of 120 days, immature hepatic uptake and conjugation processes, and increased enterohepatic circulation (4). Neonatal hyperbilirubinemia can occur due to bilirubin overproduction, decreased bilirubin conjugation, or impaired bilirubin excretion. Physiological jaundice is common both in preterm and in full term babies and occurs due to decreased bilirubin conjugation. Due to hepatic immaturity, there is a temporary deficiency of glucuronyl transferase enzymes, which reduces the rate of bilirubin conjugation resulting in a consequent retention of unconjugated bilirubin, however, it should not be confused with hereditary glucose-6-phosphate dehydrogenase deficiency. In full term infants the jaundice appears after the first 24 hours of life and reaches a peak on the 4th or 5th day while in preterm infants it usually begins 48 hours after 4th day of birth and may last up to two weeks. The average total serum bilirubin level generally ranges from 5 to 6 mg/dL on the 3rd to 4th day of life, gradually declining over the first week after birth (1). However, serum bilirubin may reach up to 260 and 360 µmol/L with < 2 mg/dL (34 μmol/L) of the conjugated form by 2nd or 3rd week of life in breast fed infants and may be asymptomatic. The levels gradually fall after a static phase of 3-4 weeks, and become normal by 4-16 weeks with continued breast feeding. In hepatitis-related jaundice, levels of conjugated bilirubin are high. Overall, a prolonged jaundice for >10 days should be thoroughly investigated.
Physiologic jaundice is caused by a combination of increased bilirubin production secondary to accelerated destruction of erythrocytes, decreased excretory capacity secondary to low levels of ligandin in hepatocytes, and low activity of the bilirubin-conjugating enzyme uridine diphosphoglucuronyltransferase (UDPGT).
Diagnosis
The diagnosis is established by examining the infant in a well-lit room and blanching the skin with digital pressure to reveal the color of the skin and subcutaneous tissue. During the examination, pathological jaundice should be ruled out. Physiological jaundice in healthy term newborns is characterized by an average total serum bilirubin level usually reaching 5 to 6 mg/dL (86 to 103 μmo/L) on the 3rd to 4th day of life and then declining over the first week after birth (1). However, there are chances that bilirubin can go up to 12 mg/dL, with less than 2 mg/dL (34 μmol/L) of the conjugated form. Maisels et al proposed criteria that can be used to exclude the diagnosis of physiologic jaundice as given in Table 1 (5).
Intrauterine Bilirubin Metabolism
Bilirubin appears in normal amniotic fluid after about 12 weeks of gestation, but it disappears by 36 to 37 weeks' gestation. Fetal liver is immature in conjugatory mechanism and removal of bilirubin from circulation. Between 17 and 30 weeks of gestation, uridine diphosphoglucuronosyl transferase (UDPGT) activity in fetal liver is only 0.1% of adult values, but it increases tenfold to 1% of adult values between 30 and 40 weeks' gestation. After birth, activity increases exponentially, reaching adult levels by 6 to 14 weeks' gestation. This increase is independent of gestation (6, 7). The major route of fetal bilirubin excretion is across the placenta. Because virtually all the fetal plasma bilirubin is unconjugated, it is readily transferred across the placenta to the maternal circulation, where it is excreted by the maternal liver. Thus, the newborn rarely is born jaundiced, except in the presence of severe hemolytic disease, when there may be accumulation of unconjugated bilirubin in the fetus. Conjugated bilirubin is not transferred across the placenta, and it also may accumulate in the fetal plasma and other tissues.
Bilirubin Production and Metabolism in Newborns
Bilirubin is produced at a rate of approximately 6 to 8 mg per kg per day in newborns, which is more than twice the production rate in adults and is attributed to polycythemia and increased red
blood cell turnover in newborns (8). In adults, the amount of bilirubin derived from sources other than the break-down of red cells is approximately 10-15% of the total, while in full-term infants it is 21-25%, and in premature infants, it is 30% of the total bilirubin load (9). The bilirubin production generally reaches a level similar to adults within 10 to 14 days postpartum (1). Bilirubin is the end product of the catabolism of heme. Newborns have a high rate of hemoglobin catabolism and bilirubin production because of their elevated hematocrit and red blood cell volume, and a shorter lifespan of the red blood cells. Major source of heme is degradation of senescent red blood cell (10). The formation of bilirubin from hemoglobin involves removal of the iron and protein moieties, followed by an oxidative process catalyzed by the enzyme microsomal heme oxygenase, an enzyme found in the reticuloendothelial system as well as many other tissues (11). The α- methane bridge of the heme porphyrin ring is opened and one mole of carbon monoxide (CO) and one mole of biliverdin and subsequently bilirubin are formed after each molecule of heme degradation (12). Biliverdin is reduced to bilirubin by biliverdin reductase. At this initial stage, bilirubin is lipid soluble and unconjugated (indirect-reacting). Bilirubin is a polar compound and at physiologic pH, it is insoluble in plasma and requires protein binding with albumin. After conjugation in the liver by glucuronosyltransferase to bilirubin diglucuronide (conjugated or direct-reacting), which is water soluble and eliminated by the liver and biliary tract (13- 16). If the albumin-binding sites are saturated, or if unconjugated bilirubin is displaced from the binding sites by medications (e.g., sulfisoxazole [Gantrisin], streptomycin, vitamin K), free bilirubin can cross the blood-brain barrier, which is toxic to the central nervous system (17). Some of the bilirubin may be converted back to its unconjugated form by a glucuronidase and reabsorbed by the intestine through enterohepatic absorption, which is known to be increased by breast milk (1).
Mechanism of Physiological Jaundice
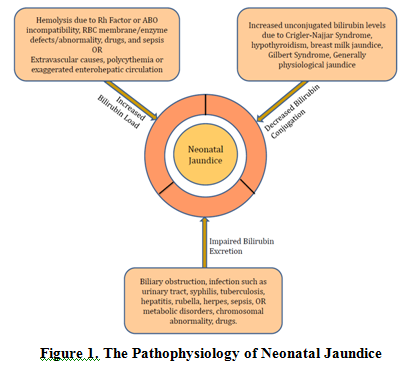
The key causes for physiological jaundice are an increased bilirubin load on liver cell, decreased hepatic uptake of bilirubin from plasma, decreased bilirubin conjugation, and defective bilirubin excretion. An overview of pathophysiology of neonatal jaundice is presented in Figure 1.
The measurement of CO in normal newborn showed that newborn produces an average of 8 to 10 mg/kg (13.7 to 17.1 μmol/ kg) of bilirubin per day (18, 19). This is more than twice the rate of normal daily bilirubin production in the adult and is explained by the fact that the neonate has a higher circulating erythrocyte volume, a shorter mean erythrocyte lifespan, and a larger early labeled bilirubin peak. Bilirubin production decreases with increasing postnatal age but is still about twice the adult rate by age 2 weeks (18). The newborn reabsorbs much larger quantities of unconjugated bilirubin by enterohepatic circulation, than does the adult. Infants have fewer bacteria in the small and large bowel and greater activity of the deconjugating enzyme β-glucuronidase (20). As a result, conjugated bilirubin, which is not reabsorbed, is not converted to urobilinogen but is hydrolyzed to unconjugated bilirubin, which is reabsorbed, thus increasing the bilirubin load on an already stressed liver. Studies in newborn humans, monkeys, and Gunn rats suggest that the enterohepatic circulation of bilirubin is a significant contributor to physiologic jaundice (21- 23). In the first few days after birth, caloric intake is low, which contributes to an increase in the enterohepatic circulation (24, 25). The decreased hepatic uptake of bilirubin from plasma is generally associated with a decreased level of ligandin. Ligandin, the bilirubin-binding protein in the human liver cell, is deficient in the liver of newborn monkeys. It reaches adult levels in the monkey by 5 days of age, coinciding with a
fall in bilirubin levels, and administration of phenobarbital increases the concentration of ligandin (26). Although this suggests that impaired uptake may contribute to the pathogenesis of physiologic jaundice, uptake does not appear to be rate limiting. A decreased uridine diphosphoglucuronosyl transferase activity is generally a cause for reduced bilirubin conjugation. Deficient UGT1A1 activity, with resultant impairment of bilirubin conjugation, has long been considered a major cause of physiologic jaundice. In human infants, the early postnatal increase in serum bilirubin appears to play an important role in the initiation of bilirubin conjugation (27). In the first 10 days after birth, UGT1A1 activity in full-term and premature neonates usually is less than 1% of adult values (12, 28). Thereafter, the activity increases at an exponential rate, reaching adult values by 6 to 14 weeks of age (12). The postnatal increase in UGT1A1 activity is independent of the infant's gestation. A defective bilirubin excretion may precipitate jaundice due to impaired excretion. The absence of an elevated serum level of conjugated bilirubin in physiologic jaundice suggests that, under normal circumstances, the neonatal liver cell is capable of excreting the bilirubin that it has just conjugated. Nevertheless, the ability of the newborn liver to excrete conjugated bilirubin and other anions (e.g., drugs, hormones) is more limited than that of the older child or adult and may become rate limiting when the bilirubin load is significantly increased. Thus, when intrauterine hyperbilirubinemia occurs, it is not uncommon to find an elevated serum level of conjugated bilirubin (5).
Pathophysiological Consequences of Hyperbilirubinemia
If the serum unconjugated bilirubin level exceeds the binding capacity of albumin, unbound lipid-soluble bilirubin crosses the blood-brain barrier though even albumin-bound bilirubin may also cross the blood-brain barrier in case of asphyxia, acidosis, hypoxia, hypoperfusion, hyperosmolality, or sepsis in the newborn (29, 30). In such a situation kernicterus may occur, resulting in neurologic consequences of the deposition of unconjugated bilirubin in brain tissue causing damage and scarring of the basal ganglia and brainstem nuclei, developmental and motor delays, sensori-neural deafness, and mild mental retardation (31). Acute bilirubin encephalopathy is caused by the toxic effects of unconjugated bilirubin on the central nervous system, and is characterized by lethargy, high-pitched cry, and poor feeding in a jaundiced infant. If acute bilirubin encephalopathy is untreated, it may progress rapidly to advanced manifestations, such as opisthotonus and seizures. It has been recommended that the bilirubin levels above 25 mg per dL (428 μmol per L) should be taken as a warning by the treating physician, however toxicity can occur at a lesser value, depending upon the genetic and ethnic conditions (32- 35). Generally, in the absence of hemolysis such risk is negligible.
Effect of Breast Feeding on Jaundice
It has been postulated that substances in maternal milk, such as β-glucuronidases, and nonesterified fatty acids may inhibit normal bilirubin metabolism, and hence may precipitate jaundice (36- 38). Further, breastfed newborns may be at increased risk for early-onset severe physiological jaundice as there is an insufficient calorie intake during first few days (39). This renders breastfed newborns at a 3-6 times higher risk of moderate-to-severe jaundice versus formula-fed newborns (39, 40). In such a scenario, breastfeeding should be continued, and if indicated formula should be added/substituted. If breastfeeding is the cause of jaundice then serum bilirubin level should decline over 48 hours (32). Certain factors present in the breast milk of some mothers may also contribute to increased enterohepatic circulation of bilirubin (breast milk jaundice). β-glucuronidase may play a role by uncoupling bilirubin from its binding to glucuronic acid, thus making it available for
reabsorption. Data suggest that the risk of breast milk jaundice is significantly increased in infants who have genetic polymorphisms in the coding sequences of the UDPGT1A1 or OATP2 genes. Although the mechanism that causes this phenomenon is not yet agreed on, evidence suggests that supplementation with certain breast milk substitutes may reduce the degree of breast milk jaundice.
Management
Although jaundice in newborns is usually benign but it should be carefully monitored, and if needed, an intervention should be given. In case of infants with mild jaundice, and phototherapy is not indicated, increasing the frequency of feedings should be advised.
Newer Concepts: Role of Oxidative Stress
Oxidative stress occurs when the production of damaging free radicals (ROS) and other oxidative molecules overwhelms the capacity of the body's antioxidant defenses, and contribute towards maintaining redox homeostasis. The initiation of stress is generally a post-natal event, however, in certain cases this can preclude such as maternal pregnancy diseases like preeclampsia, eclampsia, and maternal infections, and preterm delivery. Generally body is equipped with an array of well integrated antioxidant defenses to prevent the overage of ROS, and is available in ample quantities to scavenge and control their concentration. However, a fully efficient antioxidant defense system is lacking in preterm newborn. This may result in compromised state in pre-term neonates and renders them to complications like bronchopulmonary dysplasia, retinopathy of prematurity, hypoxic/ischemic encephalopathy, and intraventricular hemorrhage (41). The key antioxidants in human body are vitamins A, E and C, selenium, and antioxidant enzymes (catalase, superoxide dismutase, and glutathione peroxidase). It has been shown that the mean plasma total nitrite and total serum bilirubin levels and blood reticulocyte counts of the study group were significantly higher in preterm infants with newborn jaundice than those of the control group. Also, the activity of erythrocyte antioxidant enzymes and the mean plasma levels of the antioxidant vitamins A, E, and C and selenium of the preterm infants with newborn jaundice were all found to be significantly lower than those of the control group (42). Also, the jaundiced newborns had significantly lower MDA but higher SOD, catalase and GPx levels (43). Besides these key antioxidants, G6PD plays an important role in maintaining the cytosolic pool of NADPH and henceforth the cellular redox balance. Since G6PD is an important antioxidant enzyme within the erythrocytes, it is plausible that its deficiency is associated with neonatal jaundice, hemolysis and hemolytic anemia. Additionally this disruption in redox homeostasis, can lead to dysregulation of cell growth and cell signaling, resulting in abnormal embryonic development, and increase in incidence of degenerative diseases (44).
Another important antioxidant in the pathophysiology of neonatal jaundice is heme-oxygenase enzyme, which has significant activity levels in the liver, spleen, and erythropoeitic tissue. In neonates, heme-oxygenase controls production of bilirubin and hemoprotein metabolism, and maintain concentration of intracellular heme. Heme is degraded by a synergistic activity of the microsomal enzymes, heme-oxygenase and NADPH-cytochrome C (P450) reductase, and cytosolic biliverdin reductase in the presence of oxygen and NADPH, and results in production of bilirubin and carbon monoxide as by-products. Since, enzymatic activity of heme-oxygenase produces NADPH and oxygen, an up-regulation of this enzyme may overwhelm the antioxidant defenses, which include stress, poor maternal nutrition, metalloporphyrins, hormones, starvation, toxins, and xenobiotics. Additionally, it may undergo due to an increased protein synthesis and gene transcription. It has been shown that the hepatic
heme-oxygenase activity and mRNA levels are elevated in fetus and neonate as compared to adults due to an increased transcription of the heme-oxygenase gene (45). Since research indicates that many severe diseases of the neonate are caused by oxidative injury and lipid peroxidation, it is important to identify its causes, implications, and measures to minimize this.
Phototherapy and Oxidative Stress
Phototherapy is a widely used treatment modality for unconjugated hyperbilirubinemia in newborn infants due to its non-invasive nature. However, it been demonstrated that phototherapy leads to oxidative stress in preterm newborns as marked by increased markers of oxidative stress, namely lipid peroxidation and DNA damage (46). Also, phototherapy resulted in a decrease in vitamin C, uric acid, total bilirubin and MDA concentration, while there was a significant increase in the levels of total oxidant status, oxidative stress index, and lipid hydroperoxide levels, and the levels of serum total bilirubin correlated positively with MDA (47). In this regards, studies have evaluated the contribution of doses and quality of phototherapy in oxidative damage (48). In a study, where a continuous day-light phototherapy was given to jaundiced term and preterm newborns for 72 hours, levels of serum vitamin E and the activities of red blood cell anti-oxidation enzymes (superoxide dismutase, catalase and glutathione peroxidase) were measured before and after 72 h of phototherapy. The results showed that there were no changes in levels in antioxidants measured in this study. These results suggested that day-light phototherapy was safe and efficient method of treatment for all neonates presenting with hyperbilirubinemia (49). However contrasting results were observed in another study assessed the effect of phototherapy on endogenous mononuclear leukocyte DNA strand in term infants exposed to intensive or conventional phototherapy for at least 48 hours due to neonatal jaundice, and a control group. The results showed that the mean values of DNA damage scores in both the intensive and conventional phototherapy groups were significantly higher than those in the control group. Further, total oxidant status levels in both the intensive and conventional phototherapy groups were significantly higher than those in the control group. Similarly, oxidative stress index levels in both the intensive and conventional phototherapy groups were significantly higher than those in the control group. Keeping these results in view, it is suggested that both conventional phototherapy and intensive phototherapy cause endogenous mononuclear leukocyte DNA damage in jaundiced term infants (50).
Antioxidant properties of bilirubin and biliverdin
Birth is state of sudden oxidative burst marked by a sudden exposure to oxygen, resulting in high oxidative load. Since the premature newborns do not have fully efficient antioxidant defenses as maturation occurs during the late gestation period, the newborns, especially premature infants, are extremely prone to oxidative damage (51, 52). This may also impact brain in lieu of limited oxidant scavenging capacity (53, 54). In this condition, heme-oxygenase-1 is considered as highly protective in various pathophysiological states such as cardiovascular and neurodegenerative diseases owing to its reactive oxygen and nitrogen species scavenging. Further, it has been shown that direct and indirect antioxidant properties of biliverdin and bilirubin has an important role in protection of endothelial cells along with heme-oxygenase-1 (55). Besides the regular antioxidant system of glutathione redox, the bilirubin-dependent redox cycle also seems to play a role in cell protection against oxidative stress in brain. Bilirubin, a reduction product of biliverdin by biliverdin reductase, is present in brain tissue under normal conditions in nanomolar (20–50 nM) concentrations. The bilirubin-dependent redox cycle and glutathione redox cycles work hand-in-hand. Heme-
containing proteins are broken down to biliverdin by heme oxygenases (HO). As discussed previously, heme-oxygenase-1is induced by oxidative stress, and this inducible form is expressed in glial cells (55, 57). The constitutive heme-oxygenase-2 on the other hand accounts for the enzymatic activity in brain, where it is expressed in neuronal populations in several regions (58, 59). heme-oxygenase-2 impairment results in a loss of bilirubin in cells and a higher susceptibility to different CNS damages (60). It has been shown that there is a correlation between activation of heme-oxygenase-2 in cultured hippocampal and cortex neurons (59). The reduction of biliverdin to bilirubin by the cytosolic enzyme biliverdin reductase strongly induces the apoptosis of cells cultured from hippocampal/cortical structures, which is neuroprotective (61). In a recent study it was shown that bilirubin may play an antioxidant role, both in vivo and in vitro, thereby protecting the preterm infant against these oxidative stress related disorders (62, 63). The role of bilirubin is further strengthened by the finding that plasma bilirubin had a significant negative correlation with MDA but positive correlation with antioxidant enzyme activities suggesting that neonatal hyperbilirubinemia is associated with lower oxidative stress (64). Biliverdin and heme-oxygenase genes are co-expressed in brain, and biliverdin acts by exhausting free SH groups and NADH or NADPH at pH of 6.8 and 8.7, respectively (65, 66). Biliverdin is involved in cell signaling, transports the transcription factor hematin from the cytoplasm to the nucleus, allowing hematin-dependent HO-1 gene transcription (67). Silencing biliverdin leads to a depletion of cellular bilirubin, increases cellular ROS and promotes apoptotic death in neuronal cultures (68). Additionally, biliverdin increases bilirubin production from heme degradation during oxidative stress (69). An intracellularly production of bilirubin is shown to act as ROS scavenger by quenching reactive radicals before being reoxidized to biliverdin (70, 71). However, results in this regard are contradictory where infants with significant hyperbilirubinemia had elevated oxidative stress and disturbed antioxidant enzyme activity, which calls for more scientific data (72).
CONCLUSIONS
Physiological jaundice is a common condition seen in most of the newborns during their first week of life. The condition usually lasts 10 to 14 days. Here we have tried to explain the neonatal jaundice through the eyes of oxidative stress and antioxidants imbalance in neonates as an important physiological (or pathophysiological) factor.

References:
- Jaundice and hyperbilirubinemia in the newborn. In: Behrman RE, Kliegman RM, Jenson HB, eds. Nelson Textbook of pediatrics. 16th ed. Philadelphia: Saunders, 2000:511-28.
- Kessler A, Rosenberg HK. Sonographic approach to infants and children with jaundice. In: Lombay B, ed 1993 year book of pediatric radiology. Vol 5. Miskolc, Hungary: Central Medical Library of County Hospital, 1993; 3-22.
- Rous P and Drury DR, 1925
- Gartner LM, Herschel M. Jaundice and breastfeeding. Pediatr Clin North Am 2001;48:389-99.
- Maisels MJ, Gifford K: Normal serum bilirubin levels in thenewborn and the effect of breast-feeding. Pediatrics 78:837-843, 1986
- Kawade N, Onishi S. The prenatal and postnatal development of UDPglucuronyl transferase activity toward bilirubin and the effect of premature birth on this activity in the human liver. Biochem J 1981;196:257-260.
- Berk PD, Noyer C. Structure, formation, and sources of bilirubin and its transport in plasma. Semin Liver Dis 1994;14:325-330.
- Gartner LM, Herschel M. Jaundice and breastfeeding. Pediatr Clin North Am 2001;48:389-99.
- Vest, M.F. (1967) Studies on haemoglobin breakdown and incorporation of glycine into haem and bile pigment in the newborn. Bilirubin Metabolism (Ed. by I.A.D. Bouchier and B.H. Billing). Blackwell Scientific Publications, Oxford.
- Lightner DA, McDonagh AF. Molecular mechanisms of phototherapy for neonatal jaundice. Acc Chem Res 1984;17:417-424.
- Dennery PA, Rodgers P. Ontogeny and developmental regulation of heme oxygenase. J Perinatol 1996;16:S79-S83.
- Onishi S, Kawade N, Itoh S, et al. Postnatal development of uridine diphosphate glucuronyl transferase activity towards bilirubin and o- aminophenol in human liver. Biochem J 1979;194: 705-707.
- Dennery PA, Seidman DS, Stevenson DK. Neonatal hyperbilirubinemia. N Engl J Med 2001;344:581-90.
- Melton K, Akinbi HT. Neonatal jaundice. Strategies to reduce bilirubin-induced complications. Postgrad Med 1999;106:167-8,171-4,177-8.
- Wintrobe MM, Lee GR. Wintrobe’s Clinical hematology. 10th ed. Baltimore: Williams and Wilkins, 1999:267-89.
- Gartner LM, Herschel M. Jaundice and breastfeeding. Pediatr Clin North Am 2001;48:389-99.
- Moerschel SK and Cianciaruso LB. AAFP 2008
- Bartoletti AL, Stevenson DK, Ostrander CR, et al. Pulmonary excretion of carbon monoxide in the human infant as an index of bilirubin production. I.Effects of gestational age and postnatal age and some common neonatal abnormalities. J Pediatr 1979;94:952-955.
- Maisels MJ, Pathak A, Nelson NM, et al. Endogenous production of carbon monoxide in normal and erythroblastotic newborn infants. J Clin Invest 1971;50:1-9.
- Poland RL, Odell GB. Physiologic jaundice: the enterohepatic circulation of bilirubin. N Eng J Med 1971;284:1-6.
- Gourley GR. Pathophysiology of breast-milk jaundice. In: Polin RA, Fox WW,1eds. Fetal and neonatal physiology. Philadelphia: WB Saunders, 1998:1499.
- Gartner LM, Lee K-S, Vaisman S, et al. Development of bilirubin transport and metabolism in the newborn rhesus monkey. J Pediatr 1977;90:513.
- Nishioka T, Hafkamp AM, Havinga R, et al. Orlistat treatment increases fecal bilirubin excretion and decreases plasma bilirubin concentrations in hyperbilirubinemic gunn rats. J Pediatr 2003;143:327-334.
- Fevery J. Fasting hyperbilirubinemia: unraveling the mechanism involved. Gastroenterology 1997;113:1798-1800.
- Gärtner U, Goeser T, Wolkoff AW. Effect of fasting on the uptake of bilirubin and sulfobromophthalein by the isolated perfused rat liver. Gastroenterology 1997;113:1707-1713.
- Wolkoff AW, Goresky CA, Sellin J, et al. Role of ligandin in transfer of bilirubin from plasma into liver. Am J Physiol 1979; 236:E638.
- Rosenthal P, Blanckaert N, Cabra PM, et al. Formation of bilirubin conjugates in human newborns. Pediatr Res 1986;20:947-950.
- Kawade N, Onishi S. The prenatal and postnatal development of UDP glucuronyl transferase activity toward bilirubin and the effect of premature birth on this activity in the human liver. Biochem J 1981;196:257-260.
- Dennery PA, Seidman DS, Stevenson DK. Neonatal hyperbilirubinemia. N Engl J Med
2001;344:581-90.
- Ravel R. Clinical laboratory medicine: clinical application of laboratory data. 6th ed. St. Louis: Mosby, 1999:309-27.
- Melton K, Akinbi HT. Neonatal jaundice. Strategies to reduce bilirubin-induced complications. Postgrad Med 1999;106:167-8,171-4,177-8.
- Practice parameter: management of hyperbilirubinemia in the healthy term newborn. Pediatrics 1994;94(4 pt 1):558-62.
- Dennery PA, Seidman DS, Stevenson DK. Neonatal hyperbilirubinemia. N Engl J Med 2001;344:581-90.
- Maisels MJ, Newman TB. Kernicterus in otherwise healthy, breast-fed term newborns. Pediatrics 1995;96(4 pt 1):730-3.
- Newman TB, Maisels MJ. Evaluation and treatment of jaundice in the term newborn: a kinder, gentler approach. Pediatrics 1992;89(5 pt 1):809-18
- Gartner LM, Herschel M. Jaundice and breastfeeding. Pediatr Clin North Am 2001;48:389-99.
- Poland RL. Breast-milk jaundice. J Pediatr 1981;99: 86-8.
- Brodersen R, Herman LS. Intestinal reabsorption of unconjugated bilirubin. Lancet 1963;1:1242.
- Osborn LM, Reiff MI, Bolus R. Jaundice in the fullterm neonate. Pediatrics 1984;73:520-5.
- Schneider AP II. Breast milk jaundice in the newborn. A real entity. JAMA 1986;255:3270-4.
- Dani C, Cecchi A, Bertini G. Role of oxidative stress as physiopathologic factor in the preterm infant. [Article in English, Italian]. Minerva Pediatr. 2004 Aug;56(4):381-94.
- Turgut M, Ba?aran O, Cekmen M, Karata? F, Kurt A, Aygün AD. Oxidant and antioxidant levels in preterm newborns with idiopathic hyperbilirubinaemia. J Paediatr Child Health. 2004 Nov; 40(11):633-7.
- Kumar A, Pant P, Basu S, Rao GR, Khanna HD:Oxidative stress in neonatal hyperbilirubinemia.. J Trop Pediatr. 2007 Feb;53(1):69-71. Epub 2006 Dec 10.
- Ho HY, Cheng ML, Chiu DT. Glucose-6-phosphate dehydrogenase--from oxidative stress to cellular functions and degenerative diseases.[1] Redox Rep. 2007;12(3):109-18.
- Rodgers PA, Stevenson D K. Developmental biology of heme oxygenase. Clin Perinatol. 1990 Jun;17(2):275-91
- Gathwala G, Sharma S. Phototherapy induces oxidative stress in premature neonates. Indian J Gastroenterol. 2002 Jul-Aug;21(4):153-4.
- Aycicek A, Erel O.. Total oxidant/antioxidant status in jaundiced newborns before and after phototherapy.[1] J Pediatr (Rio J). 2007 Jul-Aug;83(4):319-22. Epub 2007 Jul 11
- Gathwala G, Sharma S. Oxidative stress, phototherapy and the neonate. Indian J Pediatr. 2000 Nov;67(11):805-8.
- Akisü M, Yilmaz D, Tüzün S, Kültürsay N. Antioxidant defense systems in newborns undergoing phototherapy. Indian J Pediatr. 1999 Sep-Oct; 66(5):651-5.
- A Erel O, Senturk H. Aycicek A, Kocyigit . Phototherapy causes DNA damage in peripheral mononuclear leukocytes in term infants. J Pediatr (Rio J). 2008 Mar-Apr;84(2):141-6. Epub 2008 Mar 18.
- Saugstad,O.D.(1989).The oxygen radical disease in neonatology. Indian J. Pediatr. 56, 585–593.
- Friel,J.K.,Friesen,R.W.,Harding,S.V., and Roberts,L.J.(2004).Evidence of oxidative stress in full-term healthy infants. Pediatr.Res. 56, 878–882
- Cooper,A.J.L.,Rosemberg,R.N.,Prusiner,S.B.DiMauro,S.,Barchi,R.L.,andKunk,L.M.(1997).“Glutathione in the brain:disorders of glutathione metabolism,”in The Molecular and Genetic Basis of Neurological Disease.edsR.N.Rosemberg,S.B.Prusiner,S.DiMauro, R. L.Barchi,andL.M.Kunk (Boston:Butterworth-Heinemann), 1195–1230.
- Ho,Y.S.,Magnenat,J.L.,Bronson,R. T.,Cao,J.,Gargano,M.,Sugawara, M., and Funk,C.D.(1997).Mice deficient incellular glutathione per- oxidase develop normally and show no increased sensitivity to hyperoxia. J Biol.Chem. 272, 16644–16651.
- Jansen T, Hortmann M, Oelze M, Opitz B, Steven S, Schell R, Knorr M, Karbach S, Schuhmacher S, Wenzel P, Münzel T and Daiber A (2010) Conversion of biliverdin to bilirubin by biliverdin reductase contributes to endothelial cell protection by heme oxygenase-1—evidence for direct and indirect antioxidant actions of bilirubin. Journal of Molecular and Cellular CardiologyVolume 49, Issue 2, August 2010, Pages 186–195
- Dwyer,B.E.,Nishimura,R.N.,and Lu,S.Y.(1995).Differential expression of heme oxygenase-1in cultured cortical neurons and astrocytes determined by the aid of a new heme oxygenase antibody.Response to oxidative stress. Brain Res.Mol. Brain Res. 30, 37–47.
- Calabrese,V.,Butterfield,D.A.,Scapagnini,G.,Stella,A.M.,and Maines,M.D.(2006).Redox regulation of heat shock protein expression by signaling involving nitric oxide and carbon monoxide: relevance to brain aging,neuro-degenerative disorders,and longevity. Antioxid.RedoxSignal. 8, 444–477
- Ewing,J.F.,and Maines,M.D.(1997). Histochemical localization of heme oxygenase-2 protein and mRNA expression in rat brain. Brain Res. Brain Res .Protoc. 1, 165–174.
- Mancuso,C.(2004).Heme oxygenase and its products in the nervous system. Antioxid. Redox Signal. 6, 878–887.
- Chen, J.,Tu,Y.,Connolly,E.C., and Ronnett,G.V.(2005).Heme oxygenase-2 protects against glutathione depletion-induced neuronal apoptosis mediated by bilirubin and cyclic GMP. Curr.Ne Ewing, J.F., and Maines, M.D.(1997). Histochemical localization of heme oxygenase-2 protein and mRNA expression in rat brain. Brain Res. Brain Res .Protoc. 1, 165–174.
- Doré, S., and Snyder, S.H.(1999). Neuroprotective action of bilirubin against oxidative stress in primary hippocampal cultures. Ann.N.Y. Acad.Sci. 890, 167–172.
- Acta Med Iran. 2012;50(3):153-63.Evaluation of the possible antioxidative role of bilirubin protecting from free radical related illnesses in neonates.
- Fereshtehnejad SM, Poorsattar Bejeh Mir K, Poorsattar Bejeh Mir A, Mohagheghi P.Sedlak,T.W.,Saleh,M.,Higginson,D.S.,Paul,B.D.,Juluri,K.R.,and Snyder,S.H.(2009).Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc.Natl.Acad.Sci.U.S.A. 106, 5171–5176.
- Kumar A, Pant P, Basu S, Rao GR, Khanna HD. Oxidative stress in neonatal hyperbilirubinemia. . J Trop Pediatr. 2007 Feb;53(1):69-71. Epub 2006 Dec 10.
- Ewing,J.F.,Weber,C.M.,andMaines,M.D.(1993).Biliverdin reductaseis heat resistant and coexpressed with constitutive and heatshock forms of hemeoxygenase in brain. J. Neurochem. 61, 1015–1023.
- Maines,M.D.,and Trakshel,G.M. (1993). Purification and characterization of human biliverdin reductase. Arch.Biochem.Biophys. 300, 320–326.
- Kapitulnik,J.,and Maines,M.D.(2009). Pleiotropic functions of biliverdin reductase:cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol. Sci. 30, 129–137.
- Baranano,D.E.,Rao,M.,Ferris,C.D., and Snyder,S.H.(2002).Biliverdin reductase: a major physiologic cytoprotectant. Proc.Natl.Acad.Sci. U.S.A. 99, 16093–16098.
- Miralem T., Hu Z., Torno,M.D., Lelli,K.M.,and Maines,M.D. (2005). Small interference RNA- mediated gene silencing of human biliverdin reductase,but not that of hemeoxygenase-1,attenuates arsenite-mediated induction of the oxygenase and increases apoptosis in 293A kidneycells. J. Biol.Chem. 280, 17084–17092.
- Nag N., Halder S., Chaudhuri R., Adhikary S., and Mazumder,S. (2009). Role of bilirubin as antioxidant in neonatal jaundice and effect of ethanol ice xtract of sweet lime peel on experimentally induced jaundice in rat. IndianJ.Biochem. Biophys. 46, 73–78.
- Baranano D.E., Rao M., Ferris C.D., and Snyder S.H.(2002).Biliverdin reductase: a major physiologic cytoprotectant. Proc.Natl.Acad.Sci. U.S.A. 99, 16093–16098.
- Davutoglu M, Guler E, Olgar S, Kurutas EB, Karabiber H, Garipardic M, Ekerbicer HC. Oxidative stress and antioxidant status in neonatal hyperbilirubinemia. Saudi Med J. 2008 Dec;29(12):1743-8.
|
 IJCRR
IJCRR 





 This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License





